
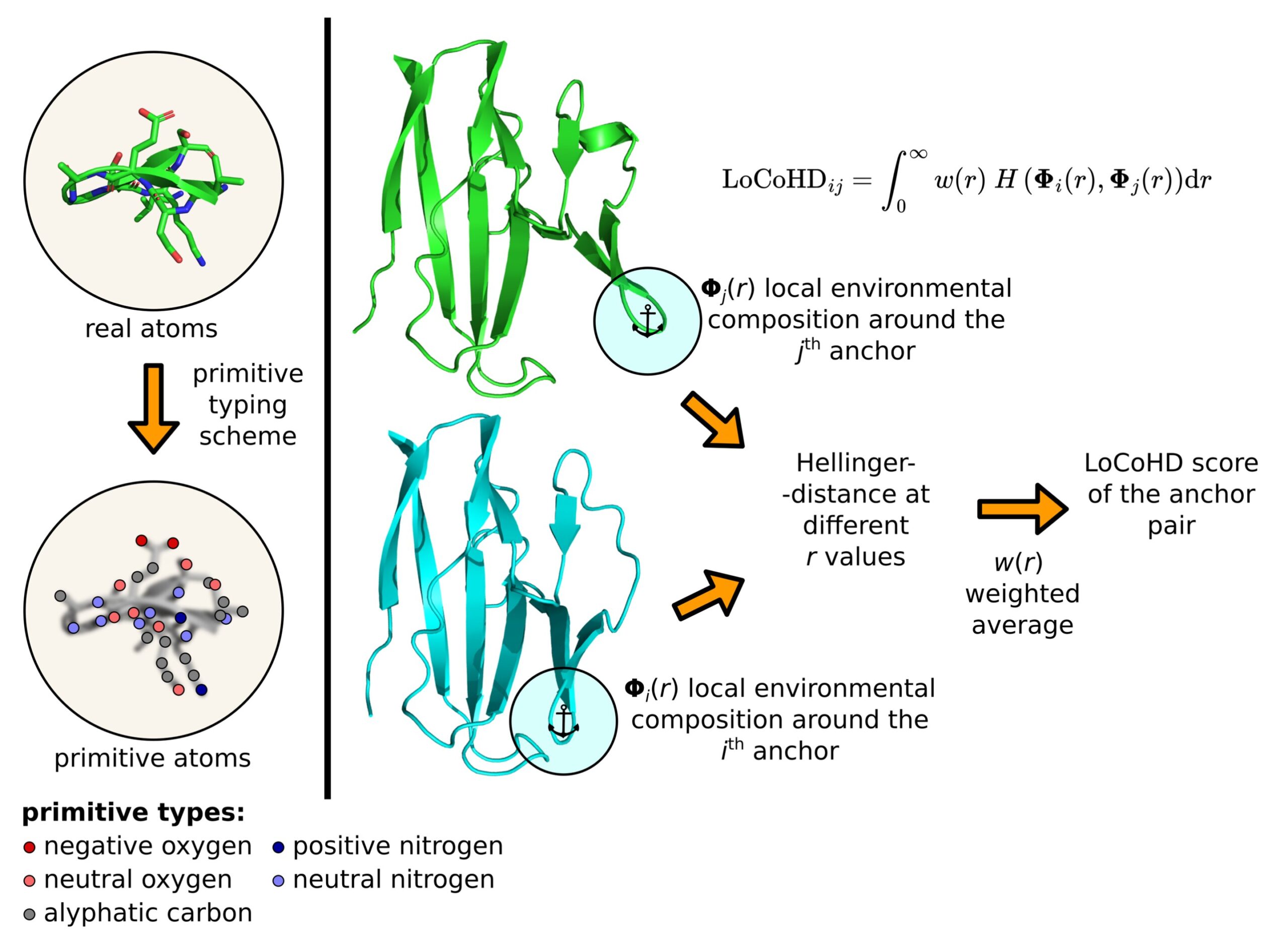
. Durch Auswahl zweier Ankeratome aus den beiden zu vergleichenden Grundstrukturen (Mitte, Grün und Türkis) kann dann mit der oben gezeigten Formel eine Zahl zwischen 0 und 1 berechnet werden. Bildnachweis: Eötvös-Loránd-Universität")
Die Abbildung zeigt den schrittweisen Prozess zur Berechnung des LoCoHD-Scores für ein bestimmtes Ankerpaar. Die chemische Auflösung erfolgt durch das primitive Typisierungsschema, mit dem die tatsächliche Struktur in eine Struktur aus primitiven Atomen umgewandelt wird (links). Durch Auswahl zweier Ankeratome aus den beiden zu vergleichenden Grundstrukturen (Mitte, Grün und Türkis) kann dann mit der oben gezeigten Formel eine Zahl zwischen 0 und 1 berechnet werden. Bildnachweis: Eötvös-Loránd-Universität
In einem kürzlich veröffentlichten Artikel in Natürliche KommunikationDie Forschungsgruppe Proteinmodellierung HUN-REN-ELTE (Institut für Chemie) hat den Grundstein für eine mathematische Methode gelegt, die einen computergestützten Vergleich der dreidimensionalen Strukturen von Proteinen ermöglicht. Das Besondere an der Methode ist, dass die bisher verfügbaren Alternativen nur die Position der Atome berücksichtigten, die neue Technik namens LoCoHD (Local Composition Hellinger Distance) jedoch auch die chemischen Informationen der Atome berücksichtigt.
Proteine sind molekulare Maschinen, die die für die Zellfunktion notwendigen Prozesse ausführen, indem sie als molekulare Schalter fungieren, Informationen aus der DNA transkribieren, kleine und große Moleküle transportieren und chemische Reaktionen im Zusammenhang mit dem Stoffwechsel regulieren. Damit das alles gelingt, muss das jeweilige Protein jedoch die richtige räumliche Konformation, also seine eigene, korrekte 3D-Anordnung haben.
Zur Bestimmung der Anordnung von Atomen in einem Protein stehen mehrere experimentelle Methoden (Röntgenkristallographie, Kernspinresonanzspektroskopie, Kryo-Elektronenmikroskopie) zur Verfügung, und in den letzten Jahrzehnten haben Proteinforscher die Form von fast 220.000 Proteinen entdeckt. Diese Ergebnisse erfordern zunehmend die Entwicklung rechnerischer Methoden, mit denen diese Anordnungen analysiert werden können.
Eine solche Methode ist der Algorithmus namens LoCoHD, der von Zsolt Fazekas, einem Ph.D., entwickelt wurde. Kandidat an der ELTE Hevesy György School of Chemistry und Forscher in der Forschungsgruppe von Dr. András Perczel. Der Algorithmus vergleicht lokale Umgebungen um Aminosäuren in Proteinen basierend auf ihrer chemischen Natur (z. B. Elementzusammensetzung, Ladung, Hydrophobie usw.).
Das Verfahren entscheidet auf einer einfachen Skala von 0 bis 1, wie unterschiedlich die betrachteten Strukturen voneinander sind. Werte nahe 0 deuten auf eine hohe Ähnlichkeit zwischen Atomanordnungen und chemischen Eigenschaften hin, während Werte nahe 1 darauf hinweisen, dass die verglichenen Proteine möglicherweise sehr unterschiedliche Eigenschaften haben. Der resultierende Zahlenwert (Metrik genannt) kann somit verwendet werden, um neue Informationen über das untersuchte System zu erhalten.
Der Algorithmus verwendet ein mehrstufiges Protokoll, um die Zahl zu generieren, die strukturelle Unterschiede darstellt. Zunächst wandelt es die echten Atome des Proteins in sogenannte primitive Atome um. Diese können als virtuell markierte Positionen dargestellt werden, deren Markierungen die chemische Natur des ursprünglichen Atoms anzeigen.
 und Strukturen (untere Felder) von His276 und Met197 in Podocin, zwei hochgradig bimodalen Aminosäuren, gemessen anhand einer Molekulardynamiksimulation. In der Simulation spielt die Aminosäure His276 eine Rolle bei der Verkürzung einer Helix, während die Aminosäure Met197 für das Füllen eines hydrophoben Hohlraums verantwortlich ist. Bildnachweis: Eötvös-Loránd-Universität")
Die Abbildung zeigt die LoCoHD-Kurven (obere Diagramme) und Strukturen (untere Felder) von His276 und Met197 in Podocin, zwei hochgradig bimodalen Aminosäuren, gemessen anhand einer Molekulardynamiksimulation. In der Simulation spielt die Aminosäure His276 eine Rolle bei der Verkürzung einer Helix, während die Aminosäure Met197 für das Füllen eines hydrophoben Hohlraums verantwortlich ist. Bildnachweis: Eötvös-Loránd-Universität
So kann ein primitives Atom beispielsweise ein „positiv geladener Stickstoff“, ein „negativ geladener Sauerstoff“, ein „neutral geladener Sauerstoff“, ein „aromatischer Kohlenstoff“ usw. sein. Die Etiketten werden nach einem sogenannten Primitivsystem generiert. Typisierungsschema, das uns in tabellarischer Form sagt, wie man reale Atome in primitive Atome umwandelt. Der Benutzer kann diese Tabelle frei spezifizieren und die chemische Auflösung der Methode festlegen.
Der zweite Schritt besteht darin, die Referenzpunkte für den Vergleich zu bestimmen, indem eine Teilmenge primitiver Atome ausgewählt wird. Diese speziell ausgewählten primitiven Atome werden Ankeratome genannt. Für jedes ausgewählte Ankeratompaar führt der Algorithmus einen Vergleichsschritt durch, dessen Ergebnis das gewünschte Unähnlichkeitsmaß ergibt. Diese Zahlen können lokal verwendet oder über einen einzelnen Deskriptor gemittelt werden, der das gesamte Protein charakterisiert.
In der Studie betonten die Forscher, dass die Methode auch bei den alle zwei Jahre stattfindenden CASP-Wettbewerben (Critical Assessment of Protein Structure Prediction), einem bekannten Wettbewerb im Bereich der Proteinforschung, eingesetzt werden kann. Bei dieser Veranstaltung verwenden die Teilnehmer verschiedene Algorithmen, um die Form von Proteinen mit bisher unbekannten Strukturen zu modellieren. CASP-Richter verwenden eine Reihe von Strukturvergleichsmethoden, um Kandidaten zu bewerten, aber keine davon berücksichtigt die Chemie der lokalen Aminosäureumgebungen.
Anhand der Daten des CASP14-Wettbewerbs 2020 führten die Forscher eine vergleichende Analyse mehrerer modellierter Proteine durch, darunter auch Strukturen, die mit der KI-basierten AlphaFold2-Methode vorhergesagt wurden. Darunter hoben sie die Analyse eines Proteins des SARS-CoV-2-Virus namens ORF8 hervor. In den modellierten Strukturen dieses Proteins wurden Aminosäureumgebungen identifiziert, die sich in ihren Interaktionsmustern deutlich von den in der experimentellen Struktur gefundenen Umgebungen unterscheiden.
Neben der Untersuchung statischer Strukturen prüften die Forscher auch, ob sich die Methode zur Analyse der inneren Bewegung von Proteinen eignet. Sie verwendeten Simulationen, die in der Lage waren, molekulare Bewegungen und aus Strukturbaugruppen extrahierte Daten zu reproduzieren. Eines der untersuchten Systeme war das Protein Podocin, das lebenswichtige Funktionen in der Niere übernimmt und dessen Mutationen schwere, oft tödliche Krankheiten verursachen können.
Die LoCoHD-Methode wurde verwendet, um Aminosäuren im Protein zu identifizieren, die während der Podocin-Bewegung großen chemischen und umweltbedingten Veränderungen unterliegen und möglicherweise sowohl seine Struktur als auch seine Funktion beeinflussen. Ebenso wurde die LoCoHD-Methode erfolgreich auf die Untersuchung des HIV-1-Kapsidproteins angewendet, bei dem eine Aminosäure identifiziert wurde, die für die Bildung der Virushülle essentiell ist.
Bei diesen Erkenntnissen handelt es sich nicht nur um Forschungskuriositäten, sondern durch eine effektivere Untersuchung von Proteinstrukturen können wir einem besseren Verständnis der Krankheitserreger, die schwere Krankheiten verursachen, und der Entwicklung wirksamer Medikamente und Behandlungen näher kommen.
Mehr Informationen:
Zsolt Fazekas et al, LoCoHD: eine Metrik zum Vergleich lokaler Proteinumgebungen, Natürliche Kommunikation (2024). DOI: 10.1038/s41467-024-48225-0
Zur Verfügung gestellt von der Eötvös-Loránd-Universität
Zitat: Neue ungarische Methode könnte der Proteinforschung helfen (29. Mai 2024), abgerufen am 29. Mai 2024 von https://phys.org/news/2024-05-hungarian-method-aid-protein.html
Dieses Dokument unterliegt dem Urheberrecht. Mit Ausnahme der fairen Nutzung für private Studien- oder Forschungszwecke darf kein Teil ohne schriftliche Genehmigung reproduziert werden. Der Inhalt dient lediglich der Information.