
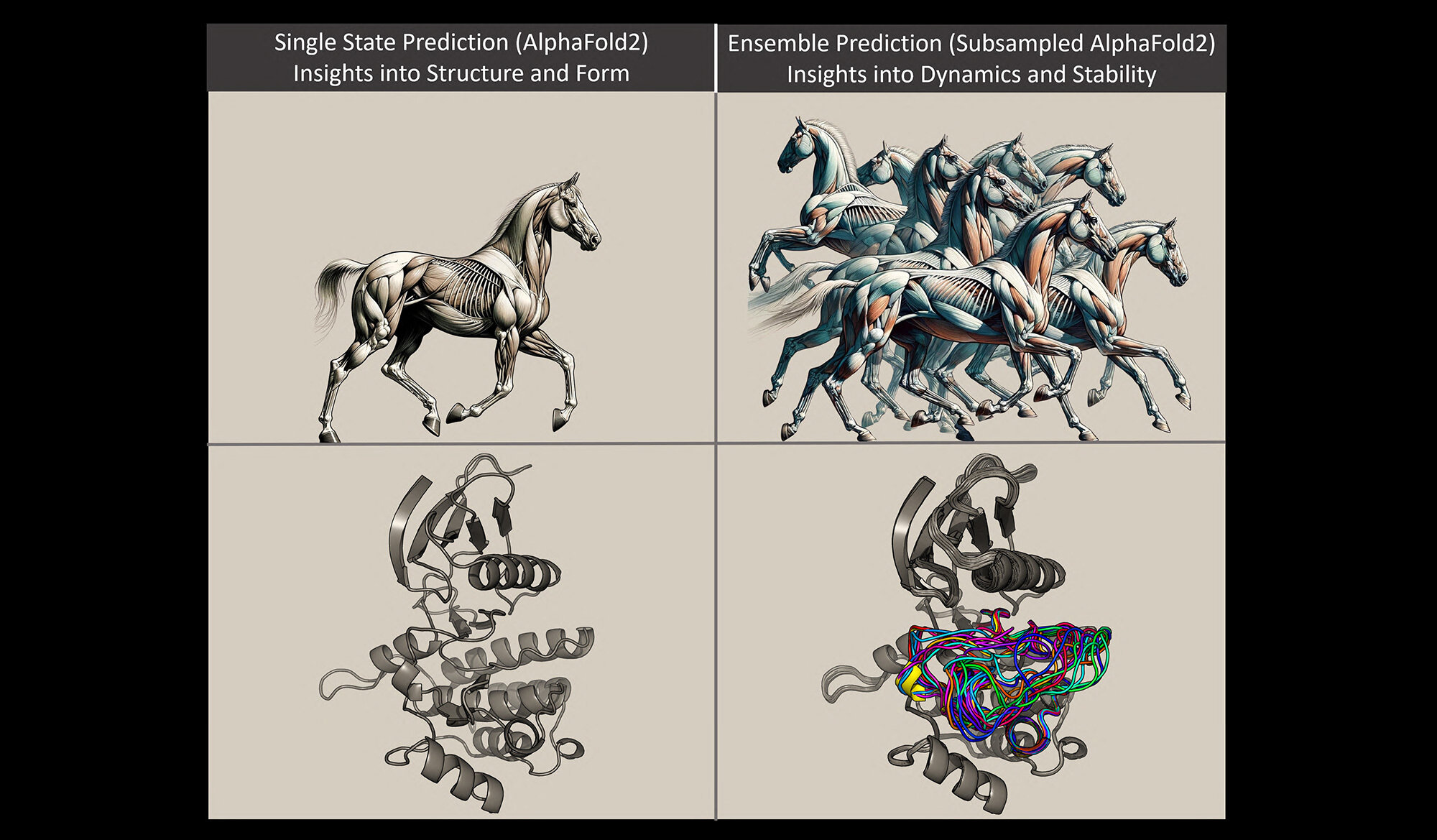

So wie mehrere Schnappschüsse eines galoppierenden Pferdes Informationen darüber liefern, wie sich das Pferd bewegt, können mehrere Schnappschüsse eines Proteins, das seine Form verändert, das wissenschaftliche Verständnis der Struktur und Funktion dieses Proteins verbessern. Bild mit freundlicher Genehmigung von Gabriel Monteiro da Silva. Bildnachweis: Gabriel Monteiro da Silva
Das Verständnis der Struktur von Proteinen ist der Schlüssel zur Entmystifizierung ihrer Funktionen und zur Entwicklung von Medikamenten, die auf sie abzielen. Zu diesem Zweck hat ein Forscherteam der Brown University eine Möglichkeit entwickelt, maschinelles Lernen zu nutzen, um schnell mehrere Proteinkonfigurationen vorherzusagen und so das Verständnis der Proteindynamik und -funktionen zu verbessern.
Eine Studie, die den Ansatz beschreibt, wurde in veröffentlicht Natürliche Kommunikation am Mittwoch, 27. März.
Die Autoren sagen, dass die Technik präzise, schnell und kosteneffektiv ist und das Potenzial hat, die Arzneimittelforschung zu revolutionieren, indem sie viel mehr Angriffspunkte für neue Behandlungen entdeckt.
Bei der gezielten Krebstherapie beispielsweise zielen Behandlungen auf Proteine ab, die das Wachstum, die Teilung und die Ausbreitung von Krebszellen steuern. Eine der Herausforderungen für Strukturbiologen bestehe darin, zelluläre Proteine gut genug zu verstehen, um Ziele zu identifizieren, sagte Studienautor Gabriel Monteiro da Silva, ein Ph.D. Kandidat für Molekularbiologie, Zellbiologie und Biochemie bei Brown.
Monteiro da Silva nutzt rechnerische Methoden zur Modellierung der Proteindynamik und sucht nach Möglichkeiten, die Methoden zu verbessern oder neue Methoden zu finden, die in verschiedenen Situationen am besten funktionieren. Für diese Studie arbeitete er mit Brenda Rubenstein, außerordentliche Professorin für Chemie und Physik, und anderen Brown-Forschern zusammen, um mit einer bestehenden KI-basierten Berechnungsmethode namens AlphaFold 2 zu experimentieren.
Während Monteiro da Silva sagte, dass die Genauigkeit von AlphaFold 2 die Vorhersage der Proteinstruktur revolutioniert habe, weist die Methode Einschränkungen auf: Sie ermöglicht es Wissenschaftlern, Proteine nur in einem statischen Zustand zu einem bestimmten Zeitpunkt zu modellieren.
„Bei den meisten zellulären Prozessen verändern Proteine ihre Form dynamisch“, sagte Monteiro da Silva.
„Um Proteinziele mit Medikamenten zur Behandlung von Krebs und anderen Krankheiten in Einklang zu bringen, benötigen wir ein genaueres Verständnis dieser physiologischen Veränderungen. Wir müssen über 3D-Formen hinausgehen, um 4D-Formen zu verstehen, wobei die vierte Dimension die Zeit ist. Das haben wir damit gemacht.“ dieser Ansatz.”
Monteiro da Silva nutzte die Analogie eines Pferdes, um Proteinmuster zu erklären. Durch die Anordnung der Muskeln und Gliedmaßen des Pferdes entstehen unterschiedliche Formen, je nachdem, ob das Pferd steht oder galoppiert; Proteinmoleküle nehmen aufgrund der Bindungsanordnungen ihrer Atombestandteile unterschiedliche Formen an.
„Stellen Sie sich vor, das Protein wäre ein Pferd“, sagte Monteiro da Silva. Frühere Methoden wurden zur Vorhersage eines stehenden Pferdemodells verwendet. Es war korrekt, aber es sagte nicht viel über das Verhalten oder Aussehen des Pferdes aus, wenn es nicht stand.
In dieser Studie konnten Forscher die Evolutionssignale des Proteins manipulieren, um mithilfe von AlphaFold 2 schnell mehrere Proteinkonformationen sowie die Häufigkeit, mit der diese Strukturen besiedelt sind, vorherzusagen.
Mithilfe der Pferdeanalogie können Forscher mit der neuen Methode schnell mehrere Schnappschüsse eines galoppierenden Pferdes vorhersagen. Das heißt, sie können sehen, wie sich die Muskelstruktur des Pferdes während seiner Bewegung verändern würde, und dann diese strukturellen Unterschiede vergleichen.
„Wenn man die vielfältigen Momentaufnahmen versteht, die die Dynamik dessen ausmachen, was mit dem Protein passiert, dann kann man mehrere verschiedene Wege finden, Proteine mit Medikamenten anzugreifen und Krankheiten zu behandeln“, sagte Rubenstein, dessen Forschung sich auf die elektronische Struktur und Biophysik konzentriert.
Rubenstein erklärte, dass das Protein, auf das sich das Team in dieser Studie konzentrierte, eines war, für das verschiedene Medikamente entwickelt wurden. Doch viele Jahre lang konnte niemand verstehen, warum bestimmte Medikamente erfolgreich waren oder scheiterten, sagte sie.
„Alles lief auf die Tatsache hinaus, dass diese spezifischen Proteine mehrere Konformationen haben, und auf das Verständnis, wie Medikamente an unterschiedliche Konformationen binden, anstatt auf die einzelne statische Struktur, die diese Techniken zuvor vorhergesagt hatten; es ist wichtig zu verstehen, wie diese Medikamente funktionieren.“ tatsächlich im Körper wirken“, sagte Rubenstein.
Beschleunigen Sie die Entdeckungszeit
Die Forscher stellten fest, dass bestehende Berechnungsmethoden teuer und zeitaufwändig sind.
„Sie sind teuer in Bezug auf Material und Infrastruktur, sie nehmen viel Zeit in Anspruch und man kann diese Berechnungen nicht wirklich mit hohem Durchsatz durchführen. Ich bin sicher, dass ich damals einer der Hauptnutzer von GPUs war.“ Brown-Computing-Cluster“, sagte Monteiro da Silva.
„In größerem Maßstab ist dies ein Problem, da es in der Welt der Proteine viel zu erforschen gibt: wie Proteindynamik und -struktur bei kaum verstandenen Krankheiten, bei Arzneimittelresistenzen und bei neu auftretenden Krankheitserregern eine Rolle spielen.“
Die Forscher beschrieben, wie Monteiro da Silva drei Jahre lang die Physik nutzte, um die Dynamik und Konformation von Proteinen zu verstehen. Mit ihrem neuen KI-basierten Ansatz konnte die Entdeckungszeit auf nur wenige Stunden reduziert werden.
„Sie können sich also vorstellen, welchen Unterschied das im Leben eines Menschen machen würde: drei Jahre statt drei Stunden“, sagte Rubenstein. „Und deshalb war es sehr wichtig, dass die von uns entwickelte Methode einen hohen Durchsatz und eine hohe Effizienz bietet.“
Was die nächsten Schritte betrifft, verfeinert das Forschungsteam seinen maschinellen Lernansatz, um ihn präziser, verallgemeinerbarer und für eine Reihe von Anwendungen nützlicher zu machen.
Mehr Informationen:
Hochdurchsatzvorhersage von Proteinkonformationsverteilungen mit unterabgetastetem AlphaFold2, Natürliche Kommunikation (2024). DOI: 10.1038/s41467-024-46715-9
Zur Verfügung gestellt von der Brown University
Zitat: Neue Technik zur Vorhersage der Proteindynamik könnte sich als großer Durchbruch für die Arzneimittelforschung erweisen (27. März 2024), abgerufen am 27. März 2024 von https://phys.org/news/2024-03-technique-protein-dynamics-big-breakthrough .html
Dieses Dokument unterliegt dem Urheberrecht. Abgesehen von der fairen Nutzung für private Studien- oder Forschungszwecke darf kein Teil ohne schriftliche Genehmigung reproduziert werden. Der Inhalt dient lediglich der Information.